We at the sickle cell and thalassemia screening programme laboratory support service receive many interesting questions from our service users.
These often fall into themes so we thought it would be useful to share some of this information with the wider diagnostic laboratory community via the PHE Screening blog.
The first theme we would like to explore involves carriers of haemoglobin variants. Haemoglobin is the substance in the blood which carries oxygen around the body. Carriers are people who have inherited one gene that makes unusual haemoglobin (haemoglobin variants).
In the laboratory, the proportion of the total haemoglobin that a variant constitutes is usually measured using high performance liquid chromatography (HPLC). We are often asked why this proportion varies.
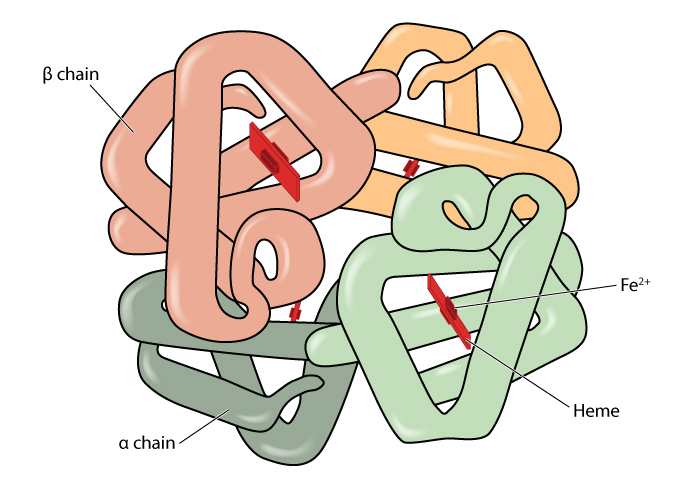
One of the most important factors that can influence the percentage of a variant is whether the amino acid substitution is situated on the alpha or beta globin chain. This is because a normal individual has two genes producing beta globin and four genes producing alpha globin.
The gene mutation producing the variant usually only affects one gene. Therefore if someone is a carrier for a beta globin variant they usually have roughly equal amounts of the variant and normal haemoglobin. If they have an alpha chain variant, only about a quarter of their haemoglobin will be affected.
However, these figures are approximate and typical percentages will depend upon the particular properties of the variant in question. For example, sickle carriers usually have approximately 40% Hb S, which is less than the 50% that would be expected for a beta chain variant. This is thought to be because the positively charged sickle globin chains do not compete as well for the alpha chains as the normal beta chains. Other beta globin variants, such as J –Baltimore, are negatively charged andcompete well for the alpha chains and therefore have higher percentages.
Another property of a variant which can affect its final proportions is how effectively it is synthesised. A good example of this is Hb E beta globin which is produced in reduced amounts, as the mutation causes abnormal DNA splicing, so a typical carrier will only have 25-30% Hb E. The stability of a variant will also be important in that unstable globins such as Haemoglobin Koln may become degraded and appear as various smaller peaks on HPLC. In these cases the amount of degradation is likely to increase with the age of the blood specimen. Therefore widely different variant proportions may be observed. Finally, some haemoglobins, such as Hb Adana, are so unstable it is not possible to see them on HPLC.
Haemoglobinopathies are very common in some populations and many people have more than one haemoglobinopathy mutation. It is not uncommon for an individual to be a carrier for a variant and also have a thalassaemia mutation. Alpha thalassaemia mutations will produce a shortage of alpha globin, which means the differing capacities of a particular variant to compete for the alpha globin chains will be exaggerated. Consequently, in the case of a sickle cell carrier in whom the Hb S already competes less well for alpha globin, co-existing alpha thalassaemia will make this worse and the total percentage of Hb S will fall in proportion to the severity of the alpha thalassaemia. However, the opposite affect – an increase in Hb S – will be observed in a person carrying Hb S and a beta thalassaemia mutation. This is because the Hb S mutation will occupy one of the two beta globin genes and the beta thalassaemia mutation on the other gene will stop or reduce the production of normal beta globin. This depends on whether it is a beta zero or beta plus thalassaemia mutation.
A final important point to consider is whether the individual has received a blood transfusion in the preceding weeks as this can have a profound (albeit temporary) effect on the proportions of any variant.
PHE Screening blogs
PHE Screening blogs provide up to date news from all NHS screening programmes – replacing our previously published newsletters.
You can register to receive updates direct to your inbox, so there’s no need to keep checking for new blogs.